





Aggiornamenti su COVID19 – Covid 2 Hospital e Policlinico Gemelli
19.217
N. tamponi eseguiti
456
N. pazienti trattamento COVID-19 ricoverati
195
Positivi al tampone
55
Ricoveri in Terapia Intensiva
910
N. pazienti trattamento COVID-19 dimessi
1.366
Totale pazienti
COVID-19

Prenota una visita
Prenotare una visita o un esame in Servizio Sanitario Nazionale o privatamente
Prenota
Reparti
L’organizzazione di 8 Dipartimenti e 138 Unità Operative con i loro relativi riferimenti e informazioni utili
Cerca
Dona Ora!
Il tuo aiuto è importante: dona ora alla Fondazione Policlinico Universitario Agostino Gemelli IRCCS
ScopriUltime News
Vuoi approfondire le nostre news visitare l’archivio Policlinico? Leggi Tutte le news
Ematriduum, chi dona sangue dona vita

Osteonecrosi dei mascellari da farmaci anti-osteoporosi e anti-tumorali

Al via la campagna di Trenta Ore per la Vita per il Progetto HOME del Gemelli

Esperti del Gemelli in Cina per un complesso intervento di chirurgia epato-biliare.

La professoressa Evis Sala nominata socio onorario della Società Giapponese di Radiologia

Alla 54ma Giornata Mondiale della Terra presentato il Percorso Clinico Assistenziale Trauma grave

13 aprile, Giornata Nazionale Malattie Neuromuscolari. Domani meeting per fare il punto su ricerca e cura

Scoperta l’impronta digitale molecolare delle lesioni pre-tumorali del pancreas

Il Gemelli insieme a Komen Italia per la salute della donna
Il Gemelli per la Giornata nazionale per la Donazione di Organi e Tessuti
Pillole Anti COVID19
Un diario scientifico per combattere il panico con l’informazione. Leggi tutte le pillole

Reumatologia, il prestigioso Carol Nachman Preis 2022 assegnato alla professoressa Maria Antonietta D’Agostino

Oncologia del Gemelli: mai ‘chiusa per COVID’. Lavorare in sicurezza è possibile
Diabete e coronavirus, l’uno può influenzare l’andamento dell’altro. Ecco perchè

Il coronavirus sta anche nelle lacrime: i consigli degli esperti

Se le rose non profumano più e il cibo non ha più sapore, potrebbe trattarsi di COVID-19
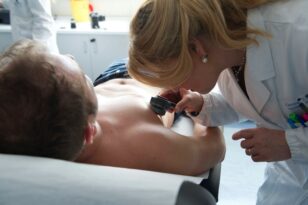
COVID-19: i dermatologi segnalano esantemi virali e ‘dita blu’ come aspetti della malattia
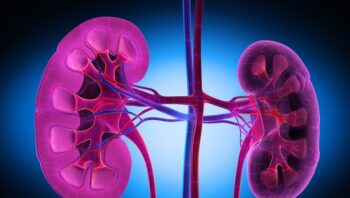
COVID-19: i rischi per i pazienti con insufficienza renale e le strategie di prevenzione
L’eparina non salverà il mondo dal COVID-19, ma se ben usata una mano ai pazienti la può dare

Oltre a farmaci e ossigeno, anche il supporto nutrizionale aiuta a superare il COVID-19

COVID-19. Gli anticorpi che “riapriranno” l’Italia. Verso la “fase 2” con la “patente di immunità”

Restiamo in cont@tto
Rimani aggiornato su news, info sulle nostre attività, open day ed eventi.
I nostri 8 Dipartimenti
Vuoi maggiori dettagli sull’organizzazione del Policlinico? Vai alla pagina






I numeri del Gemelli
1.526
Posti letto totali
94.919
Totale pazienti dimessi
400 circa
Trapianti effettuati
82.076
Accessi al Pronto Soccorso
4.110
Totale nati
53.701
Interventi Chirurgici
10.514.533
Prestazioni ambulatoriali
167.386
Prestazioni ambulatoriali interne
1.200.000
Visite al sito policlinicogemelli.it
5.322
Numero dipendenti
oltre 40
Organizzazioni Non Profit che operano internamente
62.138
Ore di formazione fruite dal personale
17,7%
Percentuale di opposizione alla donazione d’organo
2.500
Pubblicazioni scientifiche
171.297
Referti online
1.495.221
Pasti erogati a pazienti e personale
I Centri
L'eccellenza nella ricerca medica










PRENDI IL NUMERO PER LA FILA CON IL TUO SMARTPHONE
Ottimizza i tuoi tempi d'attesa con ufirst!
Controlla quante persone sono in fila, stacca il tuo biglietto e ricevi una notifica in tempo reale quando sarà il tuo turno.
Scarica subito l'app o registrati tramite il sito.


Come raggiungerci
Scopri come raggiungere il Policlinico: clicca qui
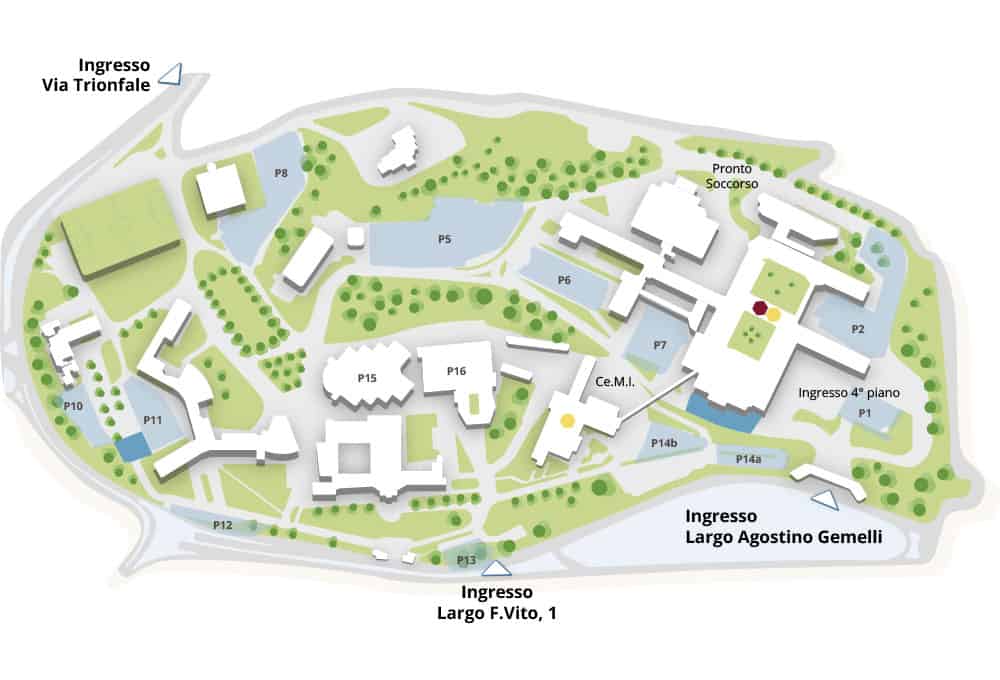
 accettazione SSN
accettazione SSN
 Accettazione Att. Privata
Accettazione Att. Privata
 parcheggi
parcheggi
 parcheggi Car Sharing *
parcheggi Car Sharing *
 ingressi
ingressi
* riservati Car2Go e ShareNow